
Introduction
A technique used for the separation of proteins based on their molecular weights is known as (SDS-PAGE) sodium dodecyl sulfate-polyacrylamide Gel electrophoresis.
SDS-PAGE Principle
The principle states that the molecules separated based on their electrophoretic mobility will migrate towards their respective electrodes when placed in an electric field. Electrophoretic mobility depends on the molecule’s structure, shape, and charge. The structure and charge of the proteins are mainly responsible for the migration. The smaller molecules will move faster due to less hindrance whereas larger ones will move slower due to greater hindrance. Thus, molecules are only separated based on their molecular weights.
The principle of SDS-PAGE is based on the separation of proteins according to their molecular weight by applying an electric field to a polyacrylamide gel matrix.
-
Electrophoretic Mobility: Molecules move in an electric field towards electrodes based on their charge-to-mass ratio. SDS-PAGE, however, neutralizes the natural charge differences between proteins.
-
Role of SDS (Sodium Dodecyl Sulfate): SDS is an anionic detergent that binds uniformly to proteins at a constant ratio (~1.4 g SDS per 1 g protein). It denatures proteins by disrupting non-covalent bonds, linearizing them and giving them a uniform negative charge. This ensures that size is the only factor affecting migration through the gel.
-
Separation by Size: Since the charge is uniform due to SDS, electrophoretic mobility depends solely on molecular size. Smaller proteins move more quickly through the pores of the polyacrylamide gel, while larger proteins face more resistance and migrate more slowly.
-
Gel Matrix: Polyacrylamide gel acts as a molecular sieve. The concentration of acrylamide determines the pore size: higher concentrations are used to resolve smaller proteins, and lower concentrations for larger proteins.
-
Reducing Conditions (optional): Often, a reducing agent like β-mercaptoethanol or DTT is added to break disulfide bonds between and within protein subunits, allowing analysis of individual polypeptides.
-
Tracking and Analysis: A tracking dye (e.g., bromophenol blue) is used to monitor the progress of the run. After electrophoresis, proteins are typically visualized using staining methods such as Coomassie Brilliant Blue or silver staining.
Material required
- Power supplies: The power supply is used to convert AC into DC.
- Gels: The gels used are either prepared in the laboratory or bought from the market in which the protein travels.
- Electrophoresis chambers: The chambers in which SDA-PAGE gels pours and protein are separated.
- Protein sample: The Protein sample used is first diluted using SDS-PAGE sample buffer, boiled for 10 minutes. Then a reducing agent is used to prevent the formation of disulfide bonds forming a tertiary structure.
- Running buffer: Buffer inside the chamber carries ions and current that maintains specific pH.
- Staining and de-staining Buffer: Stains are used to visualize the proteins
- Protein ladder: A protein ladder is used as a standard to estimate the size of the protein separated in the electrophoresis.

Agents used in SDS-PAGE
β Mercaptothion / dithiothreitol (DTT)
The disulfide bridges present between the polypeptide chains are responsible for the secondary structure of the protein. However, β Mercaptoethanol breaks the disulfide bonds that are present in the protein structure. When bonds are broken down then the S-S bonds become -SH, -SH bonds leaving the peptide end free. Therefore, on treating with β Mercaptoethanol the protein will have only a primary structure and be separated based on its size in the gel.
Sodium dodecyl sulfate (SDS)
SDS is used in the preparation of sample buffer to impart a negative charge on the protein. After the treatment with β Mercaptoethanol, all the proteins are in a linear structure so that SDS will impart a negative charge and all the proteins will move towards the positive electrode. Thus, separated only on their molecular size. Moreover, SDS is also present in the gel making sure that all the proteins should stay negative throughout the gel.
Bromophenol Blue (BPH)
Researchers use bromophenol blue as a tracking dye in the electrophoresis as it imparts color to the proteins being separated in the gel. They also mix (BPH) with a sample solution. It has a slightly negative charge so that it could reach the positive electrode before the protein molecule. It indicates the migration of protein towards the electrode.
Polyacrylamide gel
When a small amount of a cross-linker, e.g., N, N’-Methylenebisacrylamide, polymerizes with the monomer acrylamide in water, they manufacture the polyacrylamide gel. Both co-polymerize and create the 3D network of straight-chain in which diacylamine interconnects with acrylamide.

However, researchers use the other cross linkers instead of bisacrylamide:
- Piperazine diacrylate (PDA): Researchers use it to reduce silver stains in the background in SDS-PAGE.
- N, N’-diallyltartardiamide (DATD): It is a disruptable cross-linker that solubilizes the gel.
However, the ratio of acrylamide and bisacrylamide determines the size of pores in the gel. So, in a discontinuous gel system, researchers use different concentrations of pH for resolving and stacking gel.
Working of stalking gel
The interface of stacking and resolving gel separates the proteins. So, the stacking gel stacks the protein in the gel and runs separating gel approximately at the same time no matter what is the size of the protein is.
Two types of molecules are responsible for this purpose:
- Trailing Glycine molecules from tris-Glee electrophoresis buffer.
- Leading chloride ions from Tris-HCL running buffer.
When you apply the voltage, molecules migrate. Glycine is the simplest amino acid residue present in the form of zwitter-ion in stacking gel and their electrophoretic mobility is very low as compared to chloride ions. So, chloride ions will move faster as compared to the glycine residues.
In this way, chloride and glycine ions form a steep voltage gradient. Thus, proteins reside somewhere between the chloride ions (faster one) and glycine residue (slower one) and the sample becomes “stalked” into a very thin, distinct layer in order of electrophoretic mobility.

The interface between stacking gel and resolving gel
When molecules reach the interface between stacking and resolving gel the pH increases and the pore size decreases abruptly. When increases the glycine residue will no longer be in the form of zwitter-ion. They become ionized and move faster in the stacking gel. Chloride ions will travel in no time, separating the protein molecules based on their molecular sizes.
Summary of Gels
| Stacking gel | Resolving Gel | |
| Polyacrylamide concentration | Low | High |
| Pore size | larger | Smaller |
| The pH of Tris-Cl- used | 6.8 | 8.8 |
| Purpose | To stack the polypeptides on the interface of stacking gel and resolving gel. | To separate the polypeptides solely based on size. |
| Electrophoretic mobility | Glycine<protein mixture< BPB<CL– | Protein mixture<glycine<BPB<CL– |
Ammonium persulfate and TEMED
Polymerization of the gel occurs by a free radical mechanism. TEMED (N, N, N, N-tetramethyl ethylenediamine) acts as a catalyst and generates free radicals of sulfate. Then these sulfates for free radical generation are provided by ammonium persulfate
Major steps of SDS-PAGE
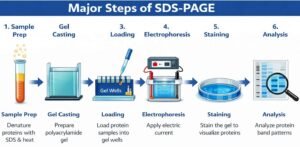
Poring of the resolving gel:
Researchers pour resolving gel between two glass plates (called one short, one tall) clipped together on a casting frame. They add isopropanol to the top of the gel to remove bubbles. (The experimenter predetermines the level of the gel by placing the comb on the glass plates and leaving approximately 1cm space below the comb. The technician uses a pen to mark the level, and they pour gel up to this mark). They allow the gel to solidify. When the gel solidifies, they remove isopropanol with filter paper.
Pouring of the stacking gel:
The solidification of resolving gel, loading of stacking takes place. You place the comb immediately after loading. You allow the gel to polymerize. After the stacking gel solidifies, you carefully remove the comb so as not to damage the well.
Loading the ladder in the wells:
Researchers add the ladder into the well very carefully using a micropipette. They mostly pre-stain the ladder with the known molecular weight protein.
Loading of samples in the well:

Researchers load each sample with an equal amount of the protein’s mixture using a micropipette. Loading of the sample should be careful, not to damage the well or sample may come out the well due to extra filling. At this stage, the sample will become blue due to (bromophenol).
Gel runs by applying the voltage:
After dipping the sandwich gel and glass plate in the running buffer, we apply voltage. We should remove the voltage when the tracking dye reaches the gel.
Subsequent analysis – (Coomassie blue staining):
The laboratory staff rinses the gel with deionized water 3-5 times to remove SDS and buffer. It may create a hindrance with the Coomassie blue stain to the proteins. Then someone shakes the gel in the blue stain in an incubator at room temperature. The invisible bands of proteins begin to appear within minutes and took almost 1 hour to complete.
Reference and Sources
- https://www.vedantu.com/biology/sds-page
- https://www.gurukpo.com/Content/model-paper-2019/science/M.Sc.%20Previous%20Year%20(BT)-%20Biological%20Macromolecules,%20Enzymology%20and%20Techniques.pdf
- https://collegedunia.com/exams/sds-page-principle-applications-limitations-and-gel-electrophoresis-biologyarticleid-3733
- https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3456489/
- https://bitesizebio.com/580/how-sds-page-works/
- https://www.coursehero.com/file/16321207/SDS-Page/
- https://www.researchgate.net/publication/224829868_Gel-Electrophoresis_and_Its_Applications
- https://www.researchgate.net/publication/269713118_Methyleneation_of_Peptides_by_N_N_N_N_-Tetramethylethylenediamine_TEMED_under_Conditions_Used_for_Free_Radical_Polymerization_A_Mechanistic_Study
- https://www.coursehero.com/file/107363059/MACUCWAKC-30306124-VPRAC2pdf/
- https://capricorn.bc.edu/bi204/wp-content/uploads/2015/08/Chapter-14-2015.pdf
- https://www.slideshare.net/rahemematthie/human-social-biology-sample-project-on-the-impact-of-heathpractices-on-the-environment
- https://www.creative-diagnostics.com/Sample-Gel-Preparation.htm
Also Read:
- Gram staining
- Proteomics: Introduction, Methods, Types and Application
- Different Staining Methods used in Microbiology
- Endospore staining and capsule staining of the bacteria
- Membrane Structure, Its Related Proteins and Hydropathy Plot
- Spectroscopy and its Types
- Enzymes: Introduction, Enzyme activity and work Mechanism
- Immunoglobulin Classes: IgG, IgA, IgM, IgD and IgE
- Different Staining Methods used in Microbiology
- Electrophoresis: Overview, Principles and Types
- Gel electrophoresis: types, principles, instrumentation and applications
- SDS-PAGE: Introduction, Principle, Working and Steps

6 thoughts on “SDS-PAGE: Introduction, Principle, Working and Steps”
Thank you so much it’s very easily understanding .👍
Very clear presentation
It’s very helpful.👍
It’s easy to understand
This post offers a really clear explanation of SDS-PAGE! I appreciate how you’ve broken down the principles and steps involved. The visuals you included also helped me grasp the concepts better. Looking forward to more detailed posts on protein analysis techniques!
This post on SDS-PAGE is incredibly informative! I especially appreciated the clear breakdown of the principles and working steps involved. It really helped me understand how protein separation works at a molecular level. Thank you for sharing such detailed insights!